Patient Information
What is HAE
Hereditary Angioedema (HAE) is a rare, chronic, potentially life-threatening genetic condition resulting from a deficiency in C1 Inhibitor protein. Estimates of the occurrence of HAE is 1 in 50,000 people with roughly the same frequency in men and women. Most patients experience their first attack during childhood or adolescence. HAE symptoms include episodes of oedema (swelling) in various body parts including the hands, feet, face, genitalia, abdomen and airway. Patients often have bouts of severe abdominal pain, nausea and vomiting that is caused by swelling in the intestinal wall. Airway swelling is particularly dangerous and can lead to death by asphyxiation. Medical help should always be sought immediately in the event of any airway swelling.
HAE patients have a defect in the gene that controls a blood protein called C1 Inhibitor. The genetic defect results in production of either inadequate or non-functioning C1-Inhibitor protein. The defective C1-Inhibitor does not perform its regulatory function of dampening the immune response and in attacks the blood vessels continue to release fluids into surrounding tissue, thereby causing oedema.
HAE is called hereditary because the genetic defect is passed on in families. A child has a 50 percent chance of inheriting this disease if one of his or her parents has it. The absence of family history does not rule out the HAE diagnosis. Many HAE cases result from patients who had a spontaneous mutation of the C1-Inhibitor gene.
Because the disease is very rare, it is not uncommon for patients to remain undiagnosed for many years. Many patients report that their frequent and severe abdominal pain was misdiagnosed. Unnecessary exploratory surgery has been performed on patients during abdominal attacks because the symptoms mimic a surgical emergency. Before therapy became available, the mortality rate for airway obstruction was reportedly as high as 30 percent.
The disorder places strain on patients, often restricting their ability to lead normal lives.
HAE Symptoms
HAE patients experience recurrent episodes of swelling in the hands, feet, face, gastrointestinal tract, genitals, and larynx (throat) that can last from two to five days. The frequency of and severity of attacks varies considerably among patients, and even among those within the same family. HAE-related swelling is not itchy or raised and is unrelieved by antihistamines and corticosteroids. About 25 percent of HAE patients experience a non-itching red rash or tingling feeling prior to an attack.
Swelling involving the feet and hands and genitalia is uncomfortable and can be extremely painful, and often prevents patients from being able to fully participate in normal daily tasks. Gastrointestinal attacks are characterised by severe abdominal pain, nausea, vomiting, and diarrhoea caused by swelling in the intestinal wall.
Laryngeal (airway) oedema is the most significant feature of HAE, because laryngeal swelling can close the airway and cause death by asphyxiation. Surveys of HAE patients reveal that approximately 50 percent experienced at least one laryngeal episode in their lives.
The development of oedema does not follow a typical pattern and therefore the site of the next episode of swelling cannot be predicted.
The frequency, duration and severity of the oedema vary considerably. Approximately a third of patients report more than one 1 attack per month; about 40% of patients experience on average 6-11 attacks per year and the remaining 22% only suffer attacks from time to time. In most cases, the symptoms of HAE increase over 12 to 36 h and last for 2-5 days before resolving. However, some patients may experience attacks that last over a week.
Most HAE attacks occur spontaneously, but patients can often link specific situations that occur in their lives, such as stress, infection, or trauma to the development of oedema.
Examples of swelling caused by HAE – WARNING! Some photos may cause distress
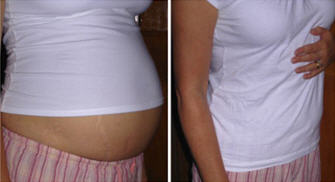
Example of an abdominal swell
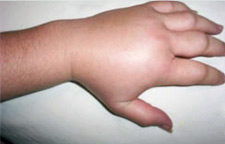
Example of a hand swell
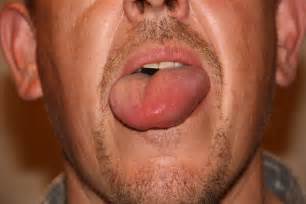
Example of a tongue swell – seek urgent medical attention
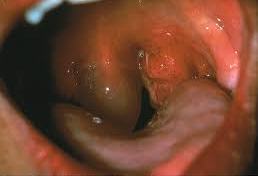
Example of laryngeal or throat swell – seek urgent medical attention
Example of internal swelling – this is swelling of the colon
HAE Diagnosis
HAE is a rare condition and can be difficult to diagnose particularly in patients where there is no family history or who only have abdominal attacks. If you have family history of HAE or suspect these are your symptoms, the first step would be to see your family doctor and then be referred on to a specialist who deals with hereditary angioedema. A diagnosis of HAE is confirmed by a blood test that confirms HAE: a screening test for C4 (complement component 4) is usually done initially and if abnormal, C1-inhibitor quantitative (antigenic) and C1- inhibitor functional levels are performed.
The most common form of the disease–Type I–is characterised by low quantitative levels of C1-inhibitor and affects about 85% of patients.
Type II HAE patients have normal or elevated levels of C1-inhibitor, but the protein does not function properly so will have abnormal functional levels.
Type III is a very rare form not due to deficiency of C1 INH. It mainly affects females and is exacerbated by high oestrogen levels. Its is becoming clear that there are rare families who have a variety of genetic defects in the complement, contact and coagulation pathways that lead to a similar presentation to classical HAE.
HAE Attack Triggers
An HAE attack occurs when the demand for C1 esterase inhibitor exceeds the amount of available functional C1 esterase in the body. Most attacks occur spontaneously, with no apparent reason; however anxiety, stress, minor trauma, surgery, and illnesses such as colds or flu can trigger attacks.
In women, menstruation and pregnancy seem to have a major effect on disease activity. Some women patients report a definite increase in the number of attacks during their menstrual periods. During pregnancy, some patients note an increase in the frequency of attacks, while others have reported a decrease.
Use of medications such as oral contraceptives and hormone replacement therapy is associated with an increase in the frequency and severity of attacks.
High blood pressure medication, known as ACE Inhibitors, have been known to increase the frequency and intensity of HAE attacks. ACE Inhibitors include Lisinopril, Fosinopril, Ramipril, Quinapril, Enalapril, Captopril, Perindopril, Trandolapril.
Treating HAE
All patients with a diagnosis of HAE should have their physician assist them in completing an ASCIA Management Plan see Management Plan(link is external)
The management of HAE consists of avoiding attack triggers, and treatment of acute attacks as well as long and short term prevention therapy as suitable for individual patients. Options are best discussed with your treating physician.
Acute Treatment
The aim of acute treatment is to halt the progression of the oedema and alleviate the symptoms. This applies particularly to episodes affecting the airways, which can cause death by suffocation.
Therapies include:
C1-INH concentrate (Berinert®) administered intravenously by a health professional in a hospital. see Berinert(link is external). Available in Australia and New Zealand.
C1_INH concentrate (Cinryze®) administered intravenously by a health professional.
Icatibant (Firazyr®) Sub Cutaneous Injection may be self administered, or given in hospital See Firazyr(link is external) Available in New Zealand.
Icatibant (Cipla) Sub Cutaneous Injection may be self administered, or given in hospital See Cipla icatibant (link is external) Available in Australia.
Preventive Treatments
Long-term prevention consists:
Lanadelumab (Takhzyro®) See Lanadelumab link is external Available in Australia
C1-INH concentrate Intravenous (Berinert®) See Berinert IV (link is external) Available in Australia and New Zealand
C1-INH concentrate Subcutaneous (Berinert®) See Berinert Subq (link is external) Available in Australia.
Danazol (Azol®) is an attenuated androgen. They can reduce the number of attacks. Because these medications are associated with a range of severe side-effects, attenuated androgens are generally reserved for patients suffering from frequent and/or severe symptoms. See Danazol(link is external)
Tranexamic acid (Cyclokapron®) the antifibrinolytic drug is used as alternative to androgens. See Tranexamic Acid(link is external)
C1-INH concentrate (Cinryze®) is an intravenous product approved for routine prevention of angioedema attacks in adults and adolescents with frequent debilitating attacks of HAE, who are intolerant to, or insufficiently protected by oral therapy. Suitability for self-administration of (Cinryze®) at home should be discussed with an immunologist.
Short-term preventative therapy is recommended for patients undergoing surgery or dental procedures which have been known to trigger an attack.
Danazol high dose androgen therapy for at least 5 days prior to surgery and 2 days afterwards.
C1-INH concentrate (Berinert® or Cinryze®) approx. 1-2 hours prior to surgery.
Support Services
HAE Australasia has a range of support services for patients and their carers specific to Australia and New Zealand.
- Elizabeth Macarthur Virtual Angioedema Centre (EMVAC)
- Meet Ups
- Healthy Mind Workshops
- Patient & Carer Conferences
- Information & Educational Resources
- Brady Club (for kids)
- Facebook Pages – HAE Australasia and HAE Australasia Support & Awareness
- Contact us
Elizabeth Macarthur Angioedema Centre (EMVAC)
Person to person virtual consultations with angioedema specialists.
EMVAC is an advisory and support service for:
- Patients with Hereditary Angioedema( HAE),
their families and carers - Interested people in the community
- Medical practitioners and nurses who provide care for HAE patients
Meet Ups
Throughout the year HAE Australasia holds informal Meet Ups, these meetings bring together patients and their carers in an informal setting over lunch or dinner where we spend time providing education, supplying resources, and offering a supportive network for patients living in that local area to have an opportunity to share experiences and gain support from one another.
Healthy Minds Workshops
HAE Australasia has engaged Clinical Psychologists to facilitate HAE Healthy Minds Workshops. These workshops have been specifically designed to give patients and carers the tools to help cope with HAE, and give patients an opportunity to discuss personal experiences in a confidential and safe environment with a mental health professional.
Upcoming Healthy Minds Workshops are listed on our Events page and via our Facebook Pages – ‘HAE Australasia’ and ‘HAE Australasia Awareness and Support‘
Patient & Carer Conferences
On each uneven year in around April or May HAE Australasia holds a Patient & Carers Conference.
The Patient & Carers Conferences are intended to bring patients, carers, physicians and industry stakeholders together in a different city each time. International and local speakers are chosen to present on the specified topic chosen for the conference.
These conferences are to provide patients and carers with education, resources, tools, and support to keep them up to date on HAE related issues.
For patients and carers living outside of the host city and state, HAE Australasia provides a travel grant to go towards your travel costs.
The next conference will be held in April 2019, more information will be released soon on our Events page and via our Facebook Pages – ‘HAE Australasia’ and ‘HAE Australasia Awareness and Support‘
Information & Educational Resources
HAE Australasia has a range of brochures and flyers to help guide you through your diagnosis.
- Information about HAE Australasia
- Someone in my family has been diagnosed with Hereditary Angioedema (HAE)
- What is HAE – information for patients, carers, families, and physicians treating HAE patients
- What’s Up with Luke? – comic book
Contact us for an information or educational pack
Brady Club (for kids)
The Brady Club is an online safe space customised for children diagnosed with HAE and their siblings.
HAE youngsters will learn how to better understand, manage and cope with their disease while offering fun ways for them to feel inspired, empowered, and have some fun! Children can play games, puzzles, whilst learning about HAE, and they can also submit stories.
Register your child for The Brady Club here
Facebook Pages
The HAE Australasia page is a closed private page for patients and their carers to ask questions, share experiences and support, and receive information, news, and updates on upcoming events.
The HAE Australasia Awareness & Support page is open to all to help raise awareness of HAE in the community. News, information and upcoming events are also posted on this page.
Contact us
We are here to help you with any enquires you have and support you may need, contact us here
DISCLAIMER: The information, including opinions and recommendations, contained in this document is for educational purposes only. It is not intended to be a substitute for professional medical advice, diagnosis or treatment. No one should act upon any information provided in this document without first seeking medical advice from a qualified, licensed medical doctor. Information derived from the internet, no matter how accurate or relevant is no substitute for competent medical care